
In brief
For decades, researchers worldwide have searched for ways to synthesize porous materials called zeolites with pore openings the right size and shape to filter out incoming molecules or to trap them and catalyze a chemical reaction. A new MIT approach should help. The challenge has been to identify a “templating molecule” to add during synthesis to create the pores needed for a targeted zeolite. Using new performance metrics and rapid atomic-level simulations, an MIT-led team evaluated nearly every molecule-zeolite pair ever proposed. The researchers then used machine learning to examine thousands of journal articles to identify pairs that had been tried experimentally. Their analyses correctly predicted zeolites formed by each molecule. Experiments with a molecule they designed yielded a zeolite that could improve catalytic converters for vehicles and factories. To help other investigators, the researchers created a publicly accessible, searchable, online database that includes their results for each molecule-zeolite combination, the articles identified by their machine learning analysis, and more.
Zeolites are a class of minerals used in everything from industrial catalysts and chemical filters to laundry detergents and cat litter. They are mostly composed of silicon and aluminum—two abundant, inexpensive elements—plus oxygen; they have a crystalline structure; and most significantly, they are porous. Among the regularly repeating atomic patterns in them are tiny interconnected openings, or pores, that can trap molecules that just fit inside them, allow smaller ones to pass through, or block larger ones from entering. A zeolite can remove unwanted molecules from gases and liquids, or trap them temporarily and then release them, or hold them while they undergo rapid chemical reactions.
Some zeolites occur naturally, but they take unpredictable forms and have variable-sized pores. “People synthesize artificial versions to ensure absolute purity and consistency,” says Rafael Gómez-Bombarelli, the Jeffrey Cheah Career Development Chair in Engineering in the Department of Materials Science and Engineering (DMSE). And they work hard to influence the size of the internal pores in hopes of matching the molecule or other particle they’re looking to capture.
The basic recipe for making zeolites sounds simple. Mix together the raw ingredients—basically, silicon dioxide and aluminum oxide—and put them in a reactor for a few days at a high temperature and pressure. Depending on the ratio between the ingredients and the temperature, pressure, and timing, as the initial gel slowly solidifies into crystalline form, different zeolites emerge.
But there’s one special ingredient to add “to help the system go where you want it to go,” says Gómez-Bombarelli. “It’s a molecule that serves as a template so that the zeolite you want will crystallize around it and create pores of the desired size and shape.”
The so-called templating molecule binds to the material before it solidifies. As crystallization progresses, the molecule directs the structure, or “framework,” that forms around it, as illustrated by the models shown below. After crystallization, the temperature is raised and the templating molecule burns off, leaving behind a solid aluminosilicate material filled with open pores that are—given the correct templating molecule and synthesis conditions—just the right size and shape to recognize the targeted molecule.

Determining the size and shape of the zeolite pores. To control the size and shape of the open pores in a zeolite, experts add a “templating molecule” that determines how the pores form as the zeolite gel solidifies. Computational methods can guide the design of a templating molecule that will produce an opening of a certain size and shape. In the models shown here, the templating molecule at the center (shown in black, white, and green) leads to the formation of the pore around it (shown in red). N+ indicates a positively charged nitrogen atom. Credit: Daniel Schwalbe-Koda, MIT
The zeolite conundrum
Theoretical studies suggest that there should be hundreds of thousands of possible zeolites. But despite some 60 years of intensive research, only about 250 zeolites have been made. This is sometimes called the “zeolite conundrum.” Why haven’t more been made—especially now, when they could help ongoing efforts to decarbonize energy and the chemical industry?
One challenge is figuring out the best recipe for making them: Factors such as the best ratio between the silicon and aluminum, what cooking temperature to use, and whether to stir the ingredients all influence the outcome. But the real key, the researchers say, lies in choosing a templating molecule that’s best for producing the intended zeolite framework. Making that match is difficult: There are hundreds of known templating molecules and potentially a million zeolites, and researchers are continually designing new molecules because millions more could be made and might work better.
For decades, the exploration of how to synthesize a particular zeolite has been done largely by trial and error—a time-consuming, expensive, inefficient way to go about it. There has also been considerable effort to use “atomistic” (atom-by-atom) simulation to figure out what known or novel templating molecule to use to produce a given zeolite. But the experimental and modeling results haven’t generated reliable guidance. In many cases, researchers have carefully selected or designed a molecule to make a particular zeolite; but when they tried their molecule in the lab, the zeolite that formed wasn’t what they expected or desired. So they needed to start over.
Those experiences illustrate what Gómez-Bombarelli and his colleagues believe is the problem that’s been plaguing zeolite design for decades. All the efforts—both experimental and theoretical—have focused on finding the templating molecule that’s best for forming a specific zeolite. But what if that templating molecule is also really good—or even better—at forming some other zeolite?
To determine the “best” molecule for making a certain zeolite framework, and the “best” zeolite framework to act as host to a particular molecule, the researchers decided to look at both sides of the pairing. Daniel Schwalbe-Koda PhD ’22, a former member of Gómez-Bombarelli’s group and now a postdoctoral fellow at Lawrence Livermore National Laboratory, describes the process as a sort of dance with molecules and zeolites in a room looking for partners. “Each molecule wants to find a partner zeolite, and each zeolite wants to find a partner molecule,” he says. “But it’s not enough to find a good dance partner from the perspective of only one dancer. The potential partner could prefer to dance with someone else, after all. So it needs to be a particularly good pairing.” The upshot: “You need to look from the perspective of each of them.”
To find the best match from both perspectives, the researchers needed to try every molecule with every zeolite and quantify how well the pairings worked.
A broader metric for evaluating pairs
Before performing that analysis, the researchers defined a new “evaluating metric” that they could use to rank each templating molecule-zeolite pair. The standard metric for measuring the affinity between a molecule and a zeolite is “binding energy,” that is, how strongly the molecule clings to the zeolite or, conversely, how much energy is required to separate the two. While recognizing the value of that metric, the MIT-led team wanted to take more parameters into account.
Their new evaluating metric therefore includes not only binding energy but also the size, shape, and volume of the molecule and the opening in the zeolite framework. And their approach calls for turning the molecule to different orientations to find the best possible fit.
Affinity scores for all molecule-zeolite pairs based on that evaluating metric would enable zeolite researchers to answer two key questions: What templating molecule will form the zeolite that I want? And if I use that templating molecule, what other zeolites might it form instead?
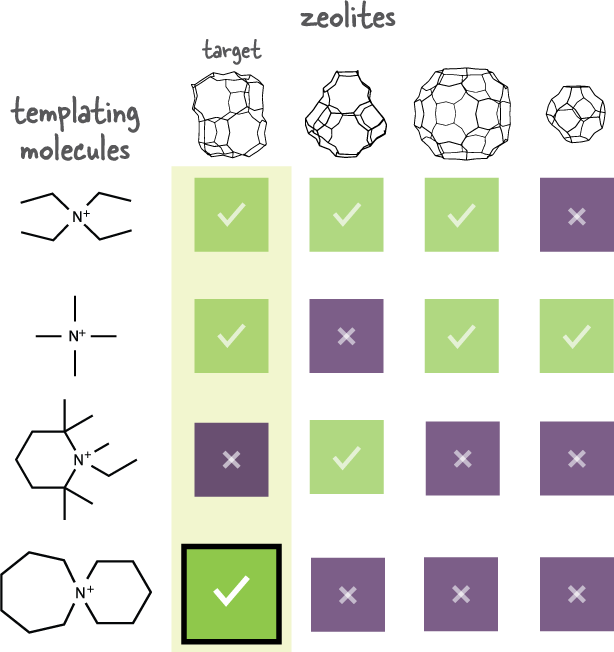
Matching the templating molecule and intended zeolite to ensure the expected outcome. This graphic illustrates the researchers’ novel approach. Using a detailed metric, they evaluate the affinity between templating molecules (listed at left) and zeolites (shown across the top); high affinity is indicated by a check, low affinity by an “X.” Using the bottom molecule to make the first zeolite (marked “target”) is a good choice as that molecule is unlikely to form anything else. Credit: Daniel Schwalbe-Koda, MIT
The figure above illustrates the process. Four zeolites are shown across the top, and four templating molecules are shown down the left side. The boxes indicate how well each pair matches up, with a check indicating a good match and an “X” a poor one. (In practice, the researchers used quantitative affinity scores in place of those qualitative indicators.) Suppose the goal is to synthesize the first zeolite from the left—marked “target.” The first and second templating molecules look like good matches for making the desired zeolite. However, they also could be good matches for making other zeolites, so they’d be a risky choice. The third molecule down wouldn’t work. But the bottom molecule is a good match for making the target zeolite and not for making any of the others. So it’s deemed “highly selective”; it’s a better, safer choice.
Validating the approach: A rich literature
But does their new metric work better than the standard one? To find out, the team needed to perform atomistic simulations using their new evaluating metric and then benchmark their results against experimental evidence reported in the literature. There are many thousands of journal articles reporting on experiments involving zeolites—in many cases, detailing not only the molecule-zeolite pairs and outcomes but also synthesis conditions and other details. Ferreting out articles with the information the researchers needed was a job for machine learning—in particular, for natural language processing.
For that task, Gómez-Bombarelli and Schwalbe-Koda turned to their DMSE colleague Elsa Olivetti PhD ’07, the Esther and Harold E. Edgerton Associate Professor in Materials Science and Engineering. Using a literature-mining technique that she and a group of collaborators had developed, she and her DMSE team processed more than 2 million materials science papers, found some 90,000 relating to zeolites, and extracted 1,338 of them for further analysis. The yield was 549 templating molecules tested, 209 zeolite frameworks produced, and 5,663 synthesis routes followed.
Based on those findings, the researchers used their new evaluating metric and a novel atomistic simulation technique to examine more than half a million templating molecule-zeolite pairs. Their results reproduced experimental outcomes reported in more than a thousand journal articles. Indeed, the new metric outperformed the traditional binding energy metric, and their simulations were orders of magnitude faster than traditional approaches.
Ready for experimental investigations
Now the researchers were ready to put their approach to the test: They would use it to design new templating molecules and try them out in experiments performed by a team led by Yuriy Román-Leshkov, the Robert T. Haslam (1911) Professor of Chemical Engineering, and a team from the Instituto de Tecnologia Química in Valencia, Spain, led by Manuel Moliner and Avelino Corma.
One set of experiments focused on a zeolite called chabazite, which is used in catalytic converters for vehicles. Using their techniques, the researchers designed a new templating molecule for synthesizing chabazite, and the experimental results confirmed their approach. Their analyses had shown that the new templating molecule would be good for forming chabazite and not for forming anything else. “Its binding strength isn’t as high as other molecules for chabazite, so people hadn’t used it,” says Gómez-Bombarelli. “But it’s pretty good, and it’s not good for anything else, so it’s selective—and it’s way cheaper than the usual ones.”
In addition, in their new molecule, the electrical charge is distributed differently than in the traditional ones, which led to new possibilities. The researchers found that by adjusting both the shape and charge of the molecule, they could control where the negative charge occurs on the pore that’s created in the final zeolite. “The charge placement that results can make the chabazite a much better catalyst than it was before,” says Gómez-Bombarelli. “So our same rules for molecule design also determine where the negative charge is going to end up, which can lead to whole different classes of catalysts.”
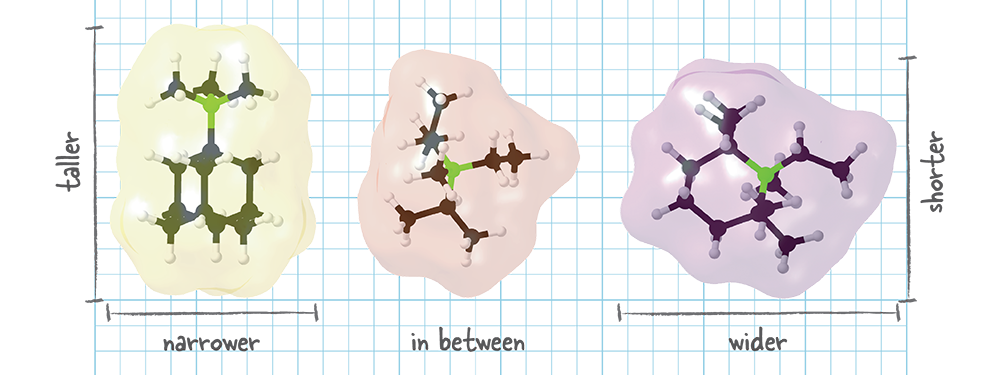
Designing a templating molecule that forms two zeolites combined in one solid. The molecule on the left is usually used to produce one zeolite, while the molecule on the right is used to produce a different one. The MIT researchers designed the templating molecule at the center, which is between the two in width and height. In experiments, the in-between molecule produced a solid that’s a combination of the two zeolites. Tests showed that the new material would be better at converting pollutants to benign compounds than the zeolite often used in catalytic converters for trucks. Credit: Daniel Schwalbe-Koda, MIT
Schwalbe-Koda describes another experiment that demonstrates the importance of molecular shape as well as the types of new materials made possible using the team’s approach. In one striking example, the team designed a templating molecule with a shape that’s halfway between the shapes of two molecules that are usually used, as illustrated in the figure above. On the left is the best-ever molecule for making chabazite, and on the right is the best-ever molecule for making a zeolite called AEI. (Every new zeolite structure is examined by the International Zeolite Association and—once approved—receives a three-letter designation.) In the center is the molecule that Schwalbe-Koda determined to be halfway between them in width and height.
Experiments using that in-between templating molecule resulted in the formation of not one zeolite or the other but a combination of the two in a single solid. “The result blends two different structures together in a way that the final result is better than the sum of its parts,” says Schwalbe-Koda. “The catalyst is like the one used in catalytic converters in today’s trucks—only better.” It’s more efficient in converting nitrogen oxides to harmless nitrogen gases and water, and—because of the two different pore sizes and the aluminosilicate composition—it works well on exhaust that’s fairly hot, as during normal operation, and also on exhaust that’s fairly cool, as during startup.
Putting the work into practice
As with all materials, the commercial viability of a zeolite will depend in part on the cost of making it. The researchers’ technique can identify promising templating molecules, but some of them may be difficult to synthesize in the lab. As a result, the overall cost of that molecule-zeolite combination may be too high to be competitive.
Gómez-Bombarelli and his team therefore include in their assessment process a calculation of cost for synthesizing each templating molecule they identified—generally the most expensive part of making a given zeolite. They use a publicly available model devised in 2018 by Connor Coley PhD ’19, now the Henri Slezynger (1957) Career Development Assistant Professor of Chemical Engineering at MIT. The model takes into account all the starting materials and the step-by-step chemical reactions needed to produce the targeted templating molecule.
However, commercialization decisions aren’t based solely on cost. Sometimes there’s a trade-off between cost and performance. “For instance, given our chabazite findings, would customers or the community trade a little bit of activity for a 100-fold decrease in the cost of the templating molecule?” says Gómez-Bombarelli. “The answer is likely yes. So we’ve made a tool that can help them navigate that trade-off.” And there are other factors to consider. For example, is this templating molecule truly novel, or have others already studied it—or perhaps even hold a patent on it?
“While an algorithm can guide development of templating molecules and quantify specific molecule-zeolite matches, other types of assessments are best left to expert judgment,” notes Schwalbe-Koda. “We need a partnership between computational analysis and human intuition and experience.”
To that end, the MIT researchers and their colleagues decided to share their techniques and findings with other zeolite researchers. Led by Schwalbe-Koda, they created an online database that they made publicly accessible and easy to use—an unusual step, given the competitive industries that rely on zeolites. The interactive website—zeodb.mit.edu—contains the researchers’ final metrics for templating molecule-zeolite pairs resulting from hundreds of thousands of simulations; all the identified journal articles, along with which molecules and zeolites were examined and what synthesis conditions were used; and many more details. Users are free to search and organize the data in any way that suits them.
Gómez-Bombarelli, Schwalbe-Koda, and their colleagues hope that their techniques and the interactive website will help other researchers explore and discover promising new templating molecules and zeolites, some of which could have profound impacts on efforts to decarbonize energy and tackle climate change.
This research involved a team of collaborators at MIT, the Instituto de Tecnologia Química (UPV-CSIC), and Stockholm University. The work was supported in part by the MIT Energy Initiative Seed Fund Program and by seed funds from the MIT International Science and Technology Initiative. Daniel Schwalbe-Koda was supported by an ExxonMobil-MIT Energy Fellowship in 2020–2021. More information about the research can be found in:
D. Schwalbe-Koda, S. Kwon, C. Paris, E. Bello-Jurado, Z. Jensen, E. Olivetti, T. Willhammar, A. Corma, Y. Román-Leshkov, M. Moliner, and R. Gómez-Bombarelli. “A priori control of zeolite phase competition and intergrowth with high-throughput simulations.” Science, vol. 374, 2021. Online: doi.org/10.1126/science.abh3350.
This article appears in the Spring 2022 issue of Energy Futures.